Sunday, February 14 2021
from the homework vault: synthesis lab

In my final college semester I took a class called Synthetic Chemistry. There were no lectures or exams. The whole class was just a set of lab experiments you'd perform at your own leisure with a partner.
I was lucky enough to be partnered with Johnny. Johnny and I were a good team. We both liked to move fast and crack jokes to keep things interesting. Some of the more diligent students took the time to complete lab write-ups as they want, but Johnny and I saved all of them for the last week of the semester. Johnny was usually looking over my shoulder while I wrote these. To get a laugh, I'd single him out in my notes by commenting on his outfit choices or ragging on his taste in indy music, knowing we'd both be pouring over these notes to finish our reports.
LinkedIn tells me Johnny went on to earn his PhD and he's currently working as a real research scientist at Tate & Lyle. Johnny - sorry, Dr. Johnny - I had so much fun that semester. Thanks for the fun memories.
January 29-30, 2013
Today I am working with Johnny on the cheese lab.
10.0168 g of powdered milk was wighed into a flask.
Added to water, heating it to 45 degrees before we add some glacial acetic acid. No diggity.
About 3.5 g of Cellite was mixed with 15 ml of 90% EtOH and filtered onto a buchner funnell.
Johnny set the cheesy cloth aside and added 1.5 K2CO3 to the water while stirring. Some bubbling. Meanwhile, I made the cellite filter and set it aside.
We put the cheese water into in an EM flask and added a bunch of boiling stones, then heating it over an open flame. A lot of bumping and foam in there, I certainly hoping those chemicals are saving themselves for marriage.
Letting it cool down. Orange, with some yellowish to it (not even), that's why I think the proteins are out. Filtered it once it was cool enough to touch. Slow. Definitely collecting some more
We stopped filtering when we were getting a lot of bubbles. Transferred it to a beaker with plenty of stones, man. Heating that up now over an open flame. Scared it's going to boil over... that would be embarrassing.
Looks ok. Not too much foaming. Wooh.
Johnny looks really good today. Goattee as always, red shirt, nice butt. I really wish I had more jeans in that color. Man alive. Johnny looks good.
The solution boiled down to about 10-15 mL. We took it off the flame and moved it to a hot plate, adding 50 mL of hot 95% EtOH and 0.4 g of decolorizing carbon. The mixture turned orange and brown. Letting it warm on a hot plate.
Meanwhile, I've made another Celite pad filter with 15 mL of EtOH and 3 g of cellite. Let it dry in a funnel. Getting ready to filter the final solution. Hoping that it will crystallize sooner than two days so we can have something to do tomorrow.
Cleaned up. Worried about the brown color. I am not expecting crystals.
Alrighty. New rainy day. I could use a nap.
Sadly the material didn't crystallize. Not only that, but the brown color is giving us the idea that it would be best to start over.
Tried again with more milk. 10.0098 g was used. Filterered out the Casein in a rag and added boiling stones to the filtrate. Heating it over an open flame. This time we used a beaker. Johnny is thinking we may have burnt the last one, giving it a brown color. I think he may be on to something.
Also, he is wearing plaid today with nicely cut jeans and boots. Excellent.
Occasional popping. White powder gathering at bottom. I don't think that's the carbonate - that would be very soluble.
Cooling it down now. It is much different than yesterday. My notes say that it was orange with yellow. This solution is clear yellow with white powder (presuming the proteins).
We poured the frothy filtrate into a big beaker with more boiling stones. Rinsed the filt flask with water and got a bit more. Heating off the water now.
New theory about the last one. I think there was too much acid. The products hydrolyzed as they were heated, yielding a final brown mixture that could only crystallize in the bizarro world.
Turned murky white when added ethanol. Nothing changed when carbon was added. Filtering it. COllecting a yellow white powder in the funnel, relinquishing a piss water kind of stuff. It's clear. I'm pumped as all hell for this solution. This is going to crystallize like a sonofabitch.
February 20, 2013
Hi there. Johnny and I are ready to begin seperating my stuff. My crude diacetylferrocene was dissolved in methylene chloride while the manual calls for it in ethyl acetate. I am now evaportating off the meth chloride. It says to use half, but I don't have a lot to begin with. I will probably just same some crude for TLC, and dissolve all of it in ehtyl acetate.
I did so. I saved a drop or two for TLC, then dissolved the rest in EtOAc. Johnny set up a really stellar column for the seperation We have six large test tubes ready to collect fractions. We are expecting resolved colored bands, so not really fretting about checking UV in the meantime.

Wow. Johnny is really good at this. We are getting beautiful separation on my cruddy diacetyl ferrocene.

The first band was yellow. Next band is orange. Above that is red that is lead a bit by yellow.

We got the second (orange) band out with a couple fractions. The top two seemed to have separated more - red and yellow. We speculated that this is 1,1' diacetyl and 1,3-diacetyl. That would be interesting insight...

After the orange band was collected, we switched from the original 20% EtOAc / Hexanes to 50:50. The red and yellow were well separated now.

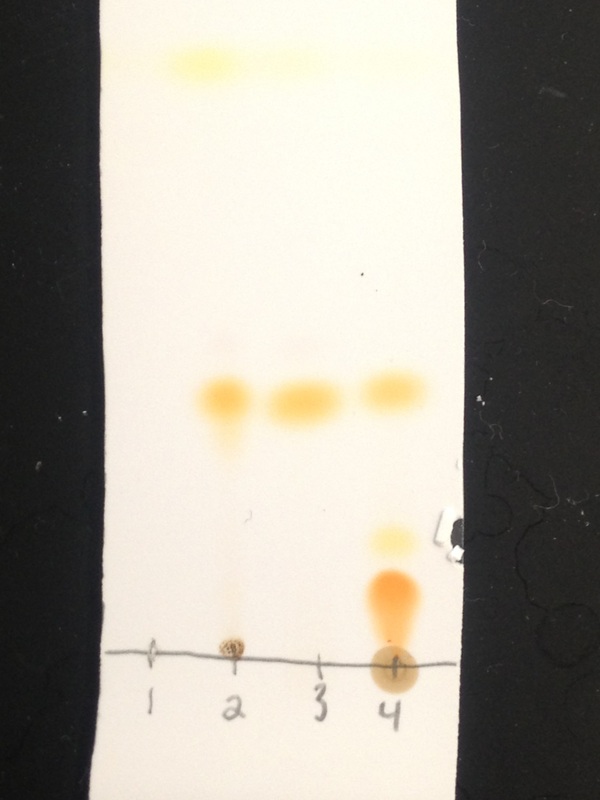
All the fractions were collected. We think the red is our diacetyl ferrocene. We are taking a TLC of the standard ferrocenes mix, Johnny's recrystallized acetylferrocene, the standard ferrocene, and the red band. Hopefully this will make it clear that the red band is indeed diacetylferrocene. There is probably not much of it, in any means. Eluting in 20% EtOAc.
I made a TLC plate with the standard mix, standard Ferrocene, J's acetylferrocene, and the red band. It matched up pretty perfectly with the first band on the standard mixture. We evaporated it on a rotary (forgot to tare it) and got a chalky red powder. Looks good. Feels good.
We will take a melting point and UV-vis next week when we start the Cholesterol lab.
February 26, 2013
We are first boiling an egg. I don't know... I guess I am just hungry, OK?

JK. It's part of the experiment! Johnny looks like he could fertilize some pretty healthy eggs on his own. Wink wink.
My yolk was 14.28 g. What a beauty. Johnny's was 14.13 g.
We combined methanol and ether (as in the manual) and mixed it for about seven minutes. Decanted it into a huge rbf flask (250 mL). Some schmutz, but no worries. Kind of took a while to decant.
Not too much recorded here. We are going to start the reflux tomorrow. Looks good. Egg gunk sitting in my drawer. We finally got it dry.

We refluxed the yellow mess in KOH / EtOH 15% wt/vol (25 mL) for 25 minutes. Dissolved easily.
Crude cholesterol weight: 0.8514 g. Crystallization in hot methanol with decolorizing carbon.
Final weight of the recrystallized cholesterol was 17.9936-17.9810
Jolly good.
March 19-20, 2013
Hey there, sailor.
Back from spring break. Dr. Niem. told us to start on the synthesis of binapthol. Starting that today.
I went a head and added 300 mL of water to a 1 L EM flask. Johnny weighed 7.2 g of 2 napthol. We combined them in the flask and started heating it.
The napthol was pale - washed purple colored and leaf like. Brittly. I have a feeling it could be a while before it dissolves.
While waiting for it to dissolve, Johnny weighed out 17 g of FeCl3. It was a big old brick. Looked like candy.
So I guess the napthol is not supposed to dissolve. Looking for oil.
Woah - there it goes. The oil formed... sort of the napthol melting. Then the whole mixture turned pink.
We added some of the iron chloride. Turned green and white. Seethes. Adding the rest over five minutes. Precipiate formed after half of it was added. Green stuff. What an incredible cauldron!

After heating it for ten minutes, we filtered out the crude green material. Looks like weed?

Isolated: Tare: 76.277 g Crude: 87.59 - tare
So we heated the crude in about 70 mL of toluene. There was a lot of crude material that didn't dissolve at boiling, so we decanted it off into a beaker and kept the material on the side. Dark solution. Let cool to room and then iced.
Well hello. When I got there, Johnny was already mixing the acetamide (sp) There was 8.96 g.
We had to make a 7.0 M solution of Bromine in acetic acid, so we measured 3.2 mL and diluted it to 9 mL with acetic acid. We slowly dripped it in over six minutes. The solution changed from brown to bright orange with ppt before I was done.
More stirring for 10 minutes.
Then poured it into cold water and filtered out the crude white material. It was white, so we did not need any NaHSO3 to consume any bromine. It was all gone. I saved some crude material for TLC analysis. It looked like about 11 g or so. Recrystallized it in two portions with ~50 mL ethanol in both.
Beautiful crystals. Not perfectly colorless, but still nice. Set out to dry. We still have to take a melting point, TLC, and NMR.
Long hairlike crystals. Beautiful... wow. Had problems with the second port. because the crystals were so fine. Just letting the funnel dry in my drawer now. It looks like we have more work to do.
I hope we have enough product.
